Evidence from genetics
Earlier in this chapter, we saw how genetic information is encoded in an organism’s DNA as a sequence of nucleobases. Over the past few decades, increasingly efficient gene-sequencing technologies have enabled geneticists to determine the precise nucleobase sequences of thousands of genes. Even whole genomesA “whole genome” sequence usually only includes protein-coding genes, not the entire DNA sequence. See the fine-print section, below, for further clarification. have been sequenced for many species, including human beings. This wealth of data has spawned a new field of research known as comparative genomics, in which the genomes of different organisms are compared using computer algorithms to search for commonalities and differences.
The claim that geneticists have sequenced “whole genomes” for many species is somewhat misleading. So-called “whole-genome sequencing” typically includes only the protein-coding genes, excluding other functional regions that often comprise much larger proportions of the organism’s DNA. For example, less than 2% of human DNA consists of protein-coding genes. A further 20% of our DNA is involved in the gene-regulatory networks that control protein-coding genes, and a total of around 80% of our DNA is active in the sense that it is transcribed to RNA, though most of its functions are unknown.For further discussion of these points, I recommend Lecture 6 of Sam Kean’s course on “Unlocking the Hidden History of DNA” in The Great Courses lecture series. The Human Genome Project, completed in 2003, succeeded in sequencing most of our protein-coding genes, but it didn’t even attempt to sequence the remaining 98% of our DNA. The ongoing ENCODE Project aims to “build a comprehensive parts list of functional elements in the human genome,” including not only protein-coding genes but also “regulatory elements that control cells and circumstances in which a gene is active.”Quotations from the ENCODE Project Overview.
As we might expect, comparisons of human DNA show that closely-related family members typically have similar genetic sequences, while distant relatives differ more. By comparing their genomes, therefore, it is possible to estimate how closely related any two people are—that is, how long ago they shared a common ancestor. Likewise, analyses comparing the genomes of different species often seem to indicate common ancestry between various kinds of organisms.
For several reasons, however, comparative genomics is not as straightforward as simply counting the number of base pairs that differ. First, the two DNA sequences must be carefully aligned to ensure that the same genes are being compared, and the alignment can be difficult when comparing genomes of different species. Since genomes typically contain large repeated segments, often they can be aligned in multiple different ways, so the comparison will depend on which alignment is chosen. To make matters worse, many genes have the mysterious ability to move around within a genome, and these so-called “jumping genes” make the alignment decisions even more challenging.Transposable elements (“jumping genes”) and other mobile genetic elements (DNA segments that can move around) constitute nearly 50% of the human genome, according to some estimates. For further discussion, I recommend Lecture 6 of Sam Kean’s course on “Unlocking the Hidden History of DNA” in The Great Courses lecture series. See also this article for discussion of the possible roles mobile genetic elements might play in the human genome. Second, there isn’t any standard way of deciding whether two sequences are similar enough to count as the “same gene” for comparison purposes. Moreover, a genome typically contains multiple copies of certain genes, and researchers have to make an arbitrary choice whether to count them as a single gene or several genes when evaluating the overall degree of similarity between genomes. Finally, and most importantly, the overall similarity between two genomes doesn’t always correlate with their relatedness. The DNA of a human boy may resemble his male cousin’s DNA more than that of his own sister, for instance, due to the major differences between the X and Y chromosomes which distinguish the sexes. For another example, the genome of an organism with polyploidy (extra copies of a chromosome resulting from a duplication mutation) may have a greater number of base pairs in common with another polyploid species than it has with non-polyploid members of its own species. Thus, careful methods of analysis must be employed to determine the true family relationships even between members of the same species, let alone to discern evolutionary relationships between different species.
A detailed examination of the methods of comparative genomics is beyond the scope of this summary, but it will be worthwhile to consider several kinds of clues typically seen as strong evidence of shared ancestry between species:For a more detailed explanation of various kinds of genetic evidence for evolution, see chapter 6 of Darrel Falk’s book Coming to Peace with Science: Bridging the Worlds Between Faith and Biology (Downers Grove: InterVarsity Press, 2004) 189-192. Falk is a Christian biologist who endorses evolutionary creationism.
- Despite the aforementioned challenges, a high degree of overall similarity between the genomes of two species usually is considered evidence that both descended from a relatively recent ancestral species. For example, human beings share more genes in common with chimpanzees than we do with any other species, and for this reason chimps are considered our closest living relatives. According to recent estimates, chimps and humans share around 94% of the same genes; some older estimates suggested that the similarity was as high as 99%.For further discussion of this example, see this article written by geologist Casey Luskin, a proponent of Intelligent Design. For a discussion of genetic evidence from an evolutionary creationist’s perspective, see chapter 6 of biologist Darrel Falk’s book Coming to Peace with Science (Downers Grove: InterVarsity Press, 2004).
No fossils have been discovered of our hypothesized common ancestor with chimps, but estimates based on comparative genomics (and studies of other related fossils) suggest that this unknown ancestral species lived between 5 and 10 million years ago. Further discussion of human origins will be undertaken in the next chapter, where we’ll consider various Christian perspectives on this topic.
- Genetic similarities are also found in pseudogenes—segments of DNA that resemble genes but do not produce functional proteins. Some pseudogenes have other functions besides producing proteins, but many appear to serve no purpose at all. In many cases, a pseudogene differs only slightly from a functional gene, suggesting that it originated as a copy of that gene but was damaged by mutation. The prevailing theory is that non-functional pseudogenes are relics of evolution: they may have served important functions for the organism’s ancestors, but they are no longer useful. The existence of identical pseudogenes in different species is considered strong evidence that the two species descended from a common ancestor. For example, in an oft-cited court case that resulted in the banning of Intelligent Design literature from a public school library, biologist Kenneth Miller (who is a Christian and an evolutionary creationist) testified that a particular pseudogene found in gorillas, chimpanzees, and human beings is compelling evidence that we descended from a common ancestor.As recorded in the trial transcript from Day 1 of Kitzmiller v. Dover Area School District (2005), Miller testified: “There’s no reason why evolution would produce a duplicate set of mistakes in two copies of things. It must mean that these two organisms are descended with modification from another organism that had the same set of mistakes. … I’d like to show you … three organisms, the gorilla, the chimpanzee, and the human being that share the exact same set of molecular mistakes.” The pseudogene identified in Miller’s testimony is named ψβ(HBBP1), and is found within a sequence of five functional β-globin genes involved in the production of the protein hemoglobin, which carries oxygen in red blood cells. Interestingly, a more recent study indicates that the pseudogene ψβ(HBBP1) does play an important role in developmental gene regulation, as we’ll see below. For further discussion of this important court case, see here and here.
- Some of the most important evidence comes from non-coding segments of DNA, called introns, found within many genes. (See the fine-print section on this page for context.) Mutations within these sequences presumedly do not affect the organism’s traits, so they are not subject to natural selection.This is a non-trivial assumption, and at least in some cases it clearly does not hold. As I explained earlier in this chapter (in the fine-print section here), the introns of one gene sometimes serve as exons for another gene. So, geneticists must be careful to avoid making such inferences regarding introns that do serve functional roles, e.g. as components of a different gene. Based on the average rates at which mutations typically accumulate, therefore, differences between introns can be used to estimate how long ago two species diverged from a common ancestor. The more time has elapsed since two nascent species became reproductively isolated from each other, the more differences we expect to find when comparing their introns. Thus, introns can serve as a sort of genetic “clock” to indicate how long ago the two new species diverged from the ancestral species.
Moreover, introns sometimes contain identifiable DNA sequences known to originate from retroviruses—viruses that trick the host cell into inserting their genetic information into the host cell’s own genome. When the viral genes are spliced into an intron, the host cell ignores the viral DNA and the host is unharmed. Nevertheless, if the insertion occurs in a germ cell (sex cell), the viral DNA can be passed to the organism’s descendants. Viral genetic sequences that are inherited in this way are called endogenous retroviruses (ERVs). Similarly, repetitive DNA segments called retroposons sometimes are inserted into introns within germ cells. These insertions, too, can be inherited by an organism’s descendants. Finding a retroposon or an ERV in the same specific location in two different genomes strongly suggests common ancestry, since it is unlikely that the same insertion would occur twice in exactly the same place just by chance. For example, the same retroposon has been found in the same intron in whales and cows, but not in horses, suggesting that cows shared a more recent common ancestor with whales than they did with horses!A retroposon named SINE-CHR-1, which is 120 nucleobases long, has been found in the same location within an intron inside the same gene in cetaceans (such as whales, dolphins, and porpoises) and even-toed ungulates (such as cows, deer, and sheep). Odd-toed ungulates, such as horses, do not share this retroposon. For further discussion of this example, see Darrel Falk’s book Coming to Peace with Science: Bridging the Worlds Between Faith and Biology (Downers Grove: InterVarsity Press, 2004) 189-192. Based on this evidence and other similar genetic indicators (along with some evidence from fossils, which we’ll discuss in the next section), evolutionary biologists have drawn the surprising conclusion that cows are indeed more closely related to whales than to horses.
Critics have responded to the above lines of evidence in a variety of ways. In rebuttal to the first point, many have noted that similar DNA doesn’t necessarily imply common descent. It may instead point to a common designer. When we find cars with similar engines and body styles, we don’t conclude that the cars descended from each other; we conclude that they were manufactured according to a similar blueprint, perhaps designed by the same team of engineers. Analogously, someone who believes that God designed all varieties of life need not be surprised to learn that He used a similar design plan for many of His creations. The degree of genetic similarity is still less surprising when we consider that DNA contains the instructions for producing many proteins and other cell parts that are common to all living organisms, regardless of how closely related those organisms may be. According to an analysis by the National Human Genome Research Institute, about 60% of human genes have a recognizable counterpart in banana plants,The corresponding banana genes are, on average, only 40% similar to ours in their precise nucleobase sequences however. See this article for further explanation. for instance; but no one thinks we’re closely related to bananas. The fact that similar organisms share many of the same genes is to be expected, whether they descended from a common ancestor or not.
On the other hand, the “common designer” rebuttal cannot easily explain why different species often share identical pseudogenes, ERVs, and retroposons. Nevertheless, there may be other ways of accounting for those similarities too. Many pseudogenes have turned out to serve important functions, including crucial roles in gene-regulatory networks. Ironically, the pseudogene Miller cited in his court testimony (see above) is one such example, as later studies revealed.The regulatory role of the pseudogene ψβ(HBBP1) is examined in Moleirinho et al., “Evolutionary Constraints in the β-Globin Cluster: The Signature of Purifying Selection at the δ-Globin (HBD) Locus and Its Role in Developmental Gene Regulation,” Genome Biology and Evolution 5 (2013), 559-571; also in Ma et al., “Genome-wide analysis of pseudogenes reveals HBBP1’s human-specific essentiality in erythropoiesis and implication in β-thalassemia,” Developmental Cell 56:4 (2021), 478-493. New functions for the non-protein-coding segments of DNA are still being discovered,See this article for further discussion. so seemingly non-functional pseudogenes may have been created for a purpose after all. Moreover, bacteria and other microbes sometimes take segments of DNA from one organism and transplant it into another. This process, called horizontal gene transfer, might account for some cases in which unrelated organisms share a few identical genes or pseudogenes. According to a recent study, at least 145 genes in the human genome appear to have come from horizontal gene transfer, and there may be others as well.Crisp, A., Boschetti, C., Perry, M. et al, “Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes,” Genome Biology 16, 50 (2015). See this article for an accessible explanation.
Alternative explanations also have been proposed for the occurrence of identical ERVs and retroposons in different species. For example, some retroviruses seem to target specific locations in a host’s genome, and this might explain why multiple species share an ERV in the same location even if they didn’t inherit it from a common ancestor. In fact, numerous cases have been identified in which identical insertions appear to have occurred independently.See this article by biologist Jonathan McLatchie for further discussion and numerous citations of mainstream biological literature acknowledging such cases. As some evolutionary geneticists have acknowledged, such cases show that shared intron sequences don’t always indicate common ancestry.For example, in this Science Daily article, evolutionary biologist Michael Lynch is quoted as saying: “Remarkably, we have found many cases of parallel intron gains at essentially the same sites in independent genotypes. This strongly argues against the common assumption that when two species share introns at the same site, it is always due to inheritance from a common ancestor.”
Another objection, frequently raised against comparative genomics as a whole, alleges that the discipline engages in circular reasoning by presupposing universal common ancestry in its methodology: comparisons usually proceed on the assumption that two species shared a common ancestor at some time in the past, and the aim of the investigation is merely to determine when and how the divergence occurred. In my opinion, this allegation doesn’t undercut the three lines of evidence listed above, but a legitimate concern may be raised as follows. Comparative genomics looks for genetic similarities to determine which organisms are closely related to each other. If these inferred relations are then cited to support the foundational assumptions of comparative genomics, there is a danger of circular reasoning here.For example, if geneticists conclude that humans and chimps are closely related because our DNA is similar, and then cite this similarity as evidence that closely-related species have similar DNA, they would be reasoning in a circle. Presumably no individual scientist would reason in such a patently fallacious way, but errors of circular reasoning might occur when scientists working in different fields of study rely on each other’s conclusions without examining the evidence that led to those conclusions. Mistakes like this might give the impression that comparative genomics provides stronger evidence for universal common descent than it really does. One may hope that other scientists would soon discover and correct these errors, but opponents of the theory of evolution often lack such faith in the scientific community.
Perhaps the biggest problem with the genetic evidence for evolution, however, has arisen from within the field of comparative genomics itself. A central goal of comparative genomics, for evolutionary science, is to construct a phylogenetic tree, also called a phylogeny—a model representing the evolutionary relationships between species, typically shown as a branching diagram, like this:
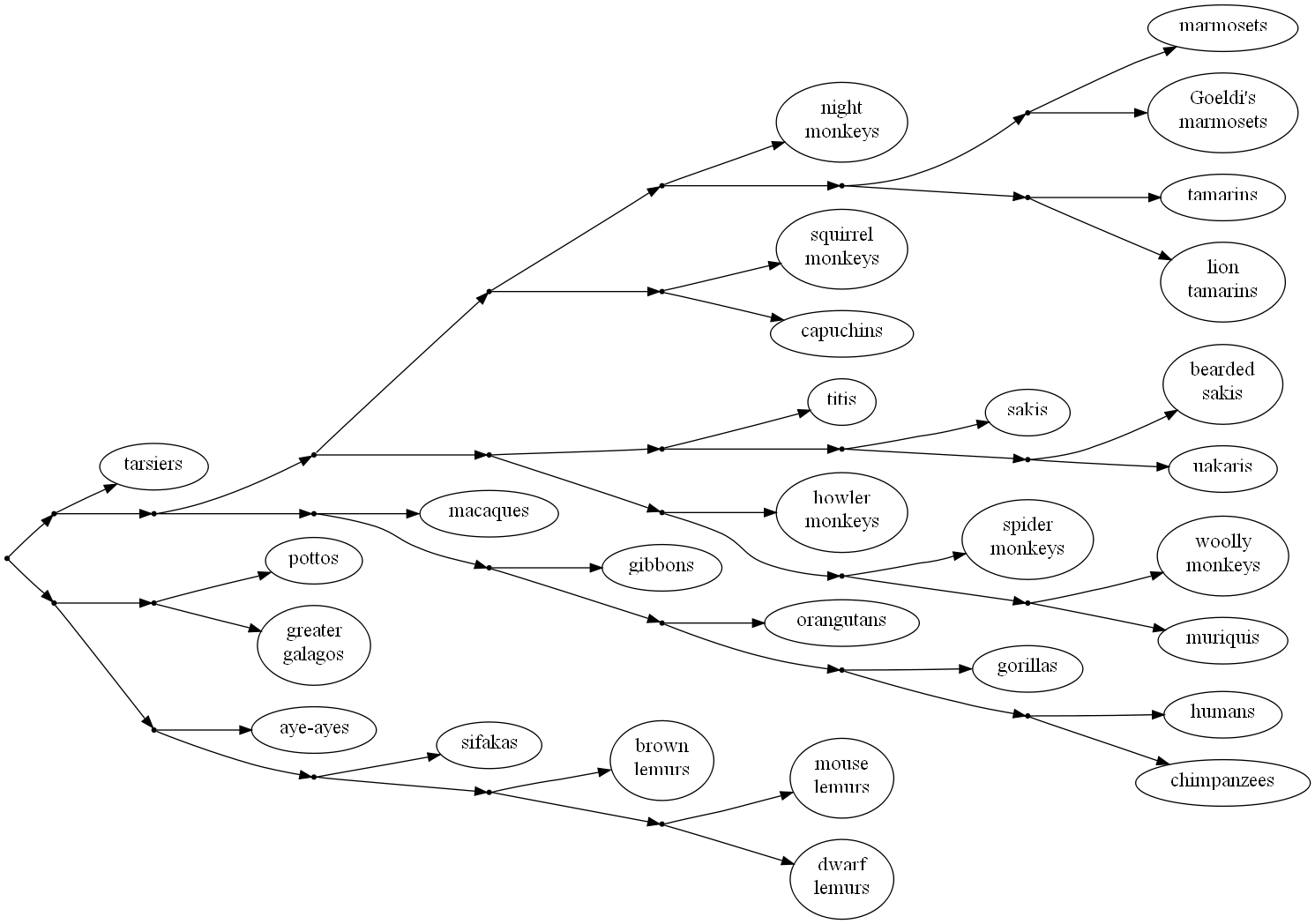
This phylogenetic tree depicts the prevailing consensus view of the evolutionary relationships among living primates. The nodes (branch points) in the diagram represent extinct ancestral species, most of which have not been discovered.
The problem is that comparisons focusing on different genes or DNA segments often yield conflicting phylogenies. For example, some portions of the human genome seem to indicate that we are more closely related to orangutans than to chimpanzees, contradicting the generally-accepted phylogeny in which orangutans are our most distant relatives among the great apes.Hobolth et. al. (2011), “Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection.” Genome research, 21(3), 349–356. For other examples and further discussion of this problem, see here and here.
Standard approaches to comparative genomics look for ways to resolve these conflicts within the framework of mainstream evolutionary theory. However, advocates of Intelligent Design have argued that conflicting phylogenies point to a different explanation for the origin of genetic information. For example, Winston Ewert argues that genetic similarities fit the pattern of a “dependency graph” used by software engineers much better than they accord with the “branching tree” structure predicted by standard evolutionary theory.Winston Ewert, “The Dependency Graph of Life,” BIO-Complexity, Vol 2018. For an accessible explanation of Ewert’s argument, see this article. In other words, according to Ewert, the evidence from comparative genomics fits the hypothesis that life was engineered better than it fits standard evolutionary theory. Although some Intelligent Design theorists do accept the doctrine of universal common descent, they argue that genetic evidence reveals an intelligently guided process of evolution. Perhaps genetic information was pre-programmed into the finely tuned laws and initial conditions of the universe, or perhaps natural evolutionary mechanisms were influenced and augmented by supernatural causes. We’ll examine some of these ideas in Chapter 11.